Quantum
Calculation on Carbon Monoxide,
Methylene Chloride and m-dichlorobenzene
Sam Isham and Stefanie Bell
Methylene Chloride and m-dichlorobenzene
Sam Isham and Stefanie Bell
Introduction
The reactivity of a molecule may be determined from the electronic structure. Molecular dipole moments, polarizability, vibrational frequencies, probability of absorption of visible light, and electron donation may be predicted from a description of the most probably locations of electrons and available energies. Molecular orbitals may be characterized by wavefunctions for electrons. Electronic structure calculations can be done by computer programs to save chemists time and money. The computer programs use different levels of theory from AM1, PM3, and ab initio used to calculate the optimized geometry, HOMO orbitals, dipole moments, and vibrational frequencies of carbon monoxide , methylene chloride, and m-dichlorobenzene. 1 The ab initio level of theory has the largest basis set and therefore gives the best geometry providing information of the potential energy surface versus bond length.
Experimental1
Carbon monoxide, methylene chloride, and m-dichlorobenzene were examined with quantum calculation programs. The program, Jmol, was used to determine optimized geometries of the AM1 and PM3 Hamiltonian’s. The results from the AM1 and PM3 were saved as “.inp” files and transferred to MacMoltPlt to be prepared for various level of theory calculations. The files were checked for complete optimization at the AM1 and PM3. MacMolplt was used to optimize the geometry at theory levels 621-G, 631-G, and DZV. Each level of theory was used as a basis set for the next best. For example, the lowest level,621-G, was based from either the AM1 or PM3 calculation. The 631-G was based from the 621-G and the DZV was based from the 631-G calculation. Each file was ran individually in GAMESS for the quantum calculation and checked for graceful exit upon completion of optimization. The bond lengths, bond angles, and HOMO and LUMO orbitals were studied in Jmol. Dipole moments were determined by opening text editor and searching for debye values. The vibrational frequencies were calculated for each molecule based from the highest level of theory: DZV. Jmol was used to create the models of bond lengths, bond angles, and HOMO and LUMO orbitals for this webpage. Each Jmol file was saved and then opened in Sea monkey to generate the webpage template. MacMolPlt was used for each ab initio level of theory of carbon monoxide to generate a potential energy surface versus bond length input file, then calculated in GAMESS, and graphed in IGOR Pro. The DZV level of theory was used for m-dichlorobenzene to write an input file for UV-Vis transition calculations. The .inp file was then calculated in GAMESS and dipole moments were searched for in the text editor upon calculation completion and recorded.
Click
the Images to view the Quantum Calculation Results for
Carbon Monoxide, Methylene Chloride, and m-dichlorobenzene
| Carbon
Monoxide |
Methylene
Chloride |
m-dichlorobenzene |
|
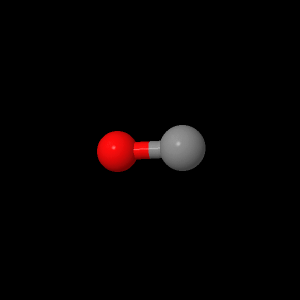 |
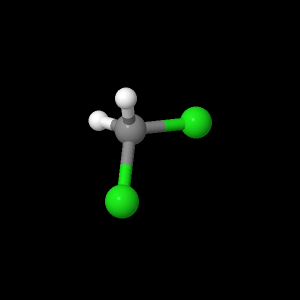 |
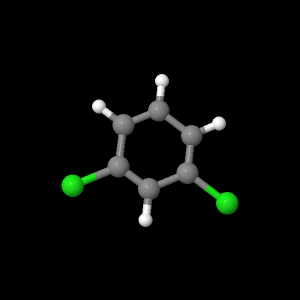 |