Introduction
The simple theories of bonding that we learn in General Chemistry are powerful and useful. These theories, which include Lewis structures, VSEPR, and hybridization, are simple models that help predict chemical properties. We should verify the usefulness of our simple predictions with molecular orbital (MO) theory. If the theoretical calculations are done carefully, we can learn a lot about chemical structure by comparing our Lewis structures and hybridization with the molecular orbitals.
The calculations in this include geometry (bond lengths,
angles), energy (molecular orbital energies), vibrational
frequencies, UV-Vis spectra, atomic charge distribution,
electrostatic potential, and dipole moment. Molecular orbital
theory is based on approximations also. These calculations are
done with some of the best available calculation methods
(Molecular Mechanics and Ab
initio). The potential energy versus bond length for
each level of theory for hydrogen fluoride was generated to show
that the bigger the basis set the better the geometry. A UV-Vis
spectrum for p-xylene was used as a comparison for calculations
of the transitions energies of the two highest levels of theory
for NHF2.
Conclusion
As mentioned in lecture, MO theory is more fundalmentally correct than VSEPR or hybridization theory and it can often allow for accurate chemical properties prediction. Although computational results usually have systematic errors, but the results were used to predict the structure and bonding of molecules. Generally, the larger the basis set the more accurate the calculation and the more computer time that is required.
| Hydrogen Fluoride (HF) |
Difluoroamine (NHF2) |
p-Xylene |
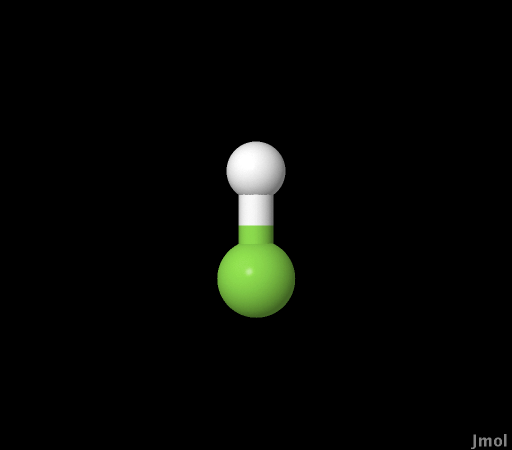 |
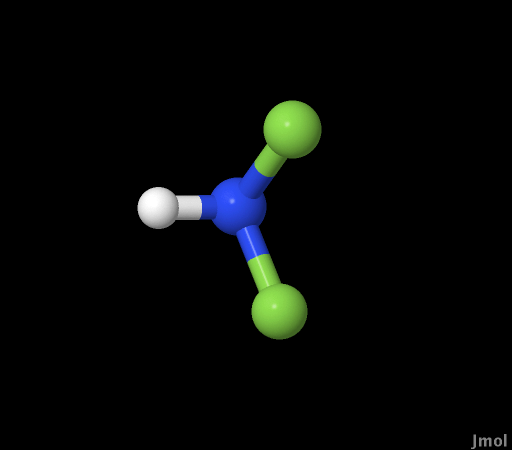 |
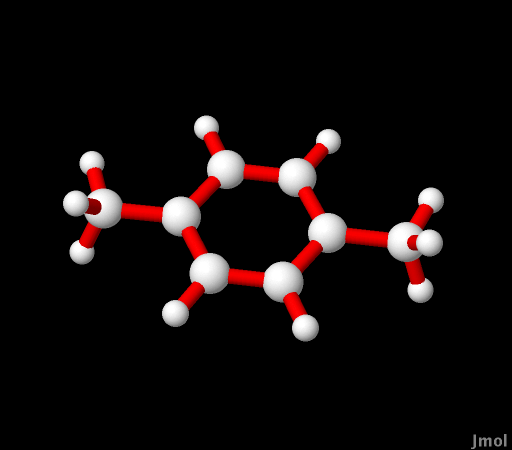 |
1. Mihalick. J; Gutow. J. Molecular Oribital Calculations. Uw Oshkosh, 2011.