Models of C8H6 were made using the best ab initio level of theory, which was double zeta valence, DZV. This is the best level of theory because it is the highest basis set of calculations run though it for the molecule.
Bond lengths of C8H6 were found using the ab initio levels.
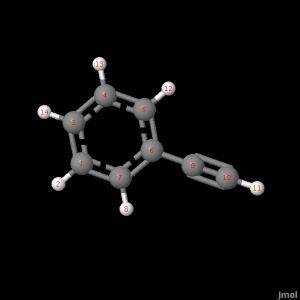
See picture about for corresponding atom numbers.
|
|
| Atom
1 |
Atom
2 |
Bond
Length
321G |
Bond
Length
Experimental1 |
| 1 |
2 |
1.072 Å | 1.087Å |
| 1 |
3 |
1.385Å | 1.397Å |
| 1 |
7 |
1.382Å | 1.393Å |
| 3 |
4 |
1.385Å | 1.397Å |
| 3 |
14 |
1.072Å | 1.087Å |
| 4 |
5 |
1.382Å | 1.393Å |
| 4 |
13 |
1.072Å | 1.087Å |
| 5 |
6 |
1.391Å | 1.408Å |
| 5 |
12 |
1.071Å | 1.074Å |
| 6 |
7 |
1.391Å | 1.408Å |
| 6 |
9 |
1.434Å | 1.430Å |
| 7 |
8 |
1.071Å | 1.087Å |
| 9 |
10 |
1.820Å | 1.210Å |
| 10 |
11 |
1.050Å | 1.066Å |
|
|
As follows is a visual model of the bond angles in phenylacetylene, with the number nearest the angle representing each's measured value.
|
|
The bond angles of C8H6 at the level of theory, 321G as well as the literature value with reference. Look to bond length photo for coresponding atom numbers.
| Atoms
|
Bond
Angle
|
Experimental
Bond
Angle1 |
| 6-9-10 |
178.956 |
180.000 |
| 6-5-12 |
119.215 |
120.114 |
| 6-7-8 |
119.662 |
139.114 |
| 9-6-7 |
119.979 |
139.740 |
| 5-6-7 |
119.450 |
119.611 |
| 5-4-13 |
119.808 |
120.779 |
| 7-1-2 |
119.900 |
120.256 |
| 4-3-1 |
120.362 |
119.843 |
| 1-7-8 |
120.091 |
100.493 |
| 3-4-13 |
120.410 |
119.821 |
| 6-5-4 |
120.355 |
119.767 |
| 6-7-1 |
120.247 |
119.767 |
| 9-6-5 |
120.571 |
119.611 |
| 9-10-11 |
197.759 |
180.000 |
| 5-4-3 |
119.777 |
119.923 |
| 7-1-3 |
119.807 |
119.923 |
| 4-5-12 |
120.429 |
120.119 |
| 4-3-14 |
119.705 |
120.079 |
| 1-3-14 |
119.932 |
120.179 |
| 3-1-2 |
120.280 |
119.821 |
The button C8H6POTENTIAL will appear in the box below.
|
|
Click below to view the phenylacetylene HOMO orbital. The HOMO orbital is the highest energy molecular orbital occupied by electrons. From the model, it can be predicted how a molecule will react.
|
|
Click below to view the phenylacetylene LUMO obital. The LUMO orbital is the lowest unoccupied molecular orbital. From the model, it can be predicted how the molecule will react.
|
|
The dipole moment for the C8H6 at the 321G basis set as well as the literature value.
| 321G |
Experimental
Value1 |
| 0.665
Debye |
0.656
Debye |
UV-Vis
Through the ab initio DZV
level of theory, the transition energy was found to be 207.802
nm. The
experimental values were found by looking at the UV-Vis
Spectrum for phenylacetylene. The values found do not match
the experimental values. For more information on phenylacetylene go to the NIST Website.
Reference
(1) U.S. Secretary of Commerce, Computational Chemistry Comparison and Benchmark
DataBase, on behalf of the United States of America, 2010. Website: <http://cccbdb.nist.gov/>
Page skeleton
and JavaScript generated by export to web function using Jmol 11.8.20 2010-02-28 19:28
on Mar 24, 2010.